
Written by Pendell Meyers and Steve Smith
Here are two cases of middle-aged men with chest pain who had prehospital ECGs.
Patient 1, ECG 1: |
| What do you think? |
Patient 2, ECG 2:

What do you think?
Queen of Hearts interpretation of ECG 1:

Queen of Hearts interpretation for ECG 2:

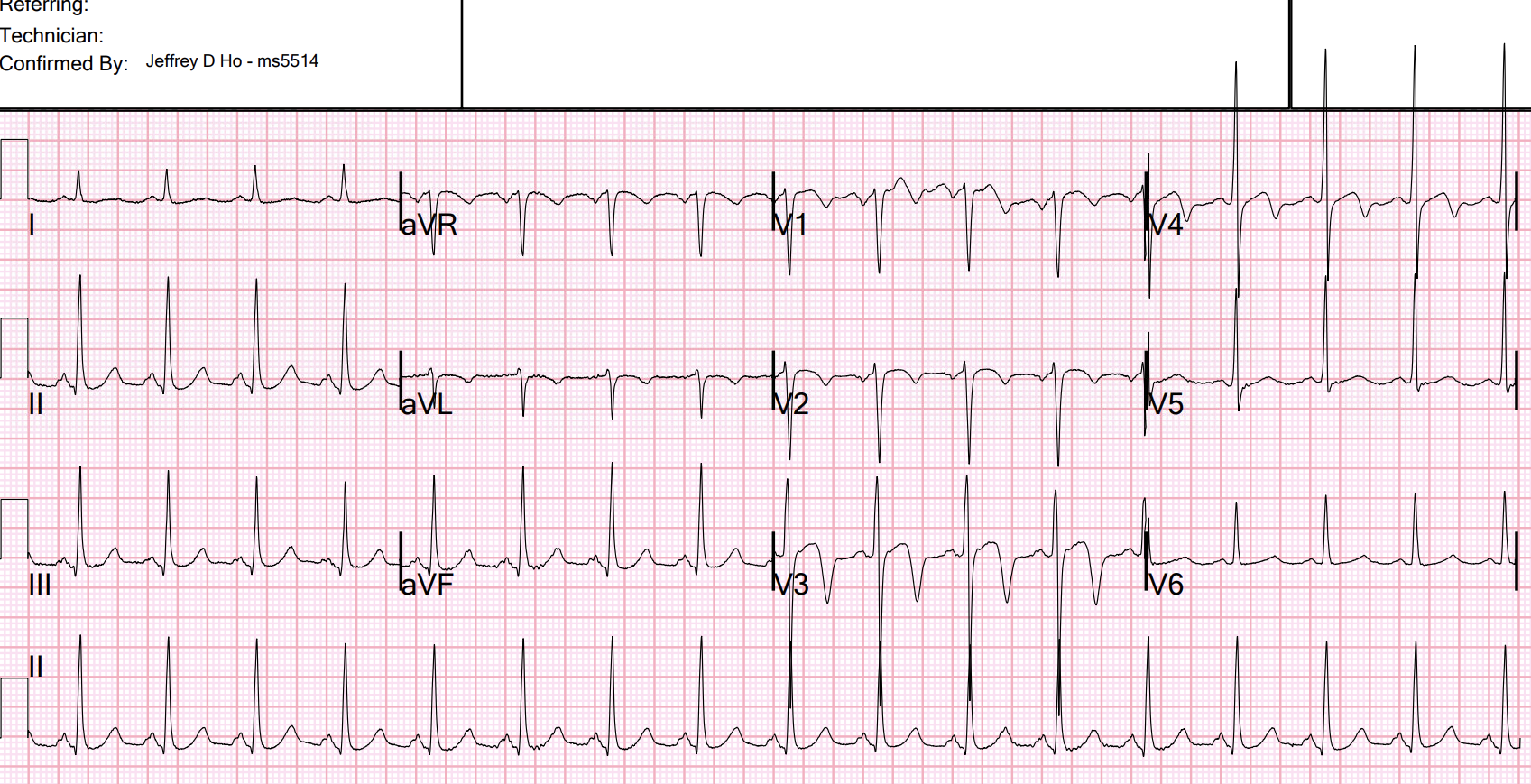
Interpretation of ECG 1 (OMI): Sinus rhythm, normal QRS, with easily diagnostic signs specific for inferior and posterior wall transmural ischemia, with the most likely etiology of course being acute coronary occlusion MI. Inferior T waves are hyperacute, with reciprocal negative hyperacute T waves in aVL. Posterior OMI is indicated by the inappropriate ST depression maximal in V2. However, I do not believe this case has sufficient STE to meet STEMI criteria.
Interpretation of ECG 2: (Not OMI): There ﹥1 mm of ST elevation in consecutive inferior leads (meets STEMI criteria), with no reciprocal ST depression in aVL. There is a slight T-wave inversion in aVL. The upward concavity of the ST segments is pronounced. The T-waves are NOT hyperacute; they do not have much "bulkiness" especially in proportion to the well-formed R-waves. This is a NORMAL ECG.
Outcome of case with ECG 2: Inferior STEMI was diagnosed by the emergency physician and the patient needed to be flown by helicopter to a cath lab. The arteries were clean. There was no MI. This was the patient's baseline ECG. It was a false positive. The patient ruled out for MI by serial troponins. Chest pain was non-cardiac.
Register for access to Queen of Hearts here
Case 1 continued:
Time = 50 minutes, in the ED:
 |
| Now it meets STEMI criteria also. Although its not labelled, V5 and V6 are likely posterior leads. |

Initial hs trop I: 56 ng/L
Time = 80 minutes
Angiogram:
Acute 100% (TIMI 0) RCA occlusion, PCI performed



Post PCI ECG:

 |
| Independently diagnostic for inferoposterior reperfusion. (deeply inverted inferior T-waves, and increased amplitude T-waves in V2, V3) Smith: Why do the T-waves in V2 and V3 enlarge in reperfusion of the posterior wall? If the recording was on the posterior wall, there would be T-wave inversion. But T-wave inversion measure from the anterior wall (opposite) appears UPRIGHT! Then add this upright deflection to the already upright T-waves of the anterior wall and you have extra large anterior T-waves!! See our paper here: Driver BE, Shroff GR, Smith SW. Posterior reperfusion T-waves: Wellens’ syndrome of the posterior wall. Emerg Med J [Internet]. 2017;34:119–123. Available from: http://dx.doi.org/10.1136/emermed-2016-205852 |
High sensitivity Troponin Ts (ng/L): 56
1,656
14,458
9,942
Echo:
EF 57%
Basal to mid inferolateral hypokinesis
Learning Points:
1. Expert or Queen of hearts interpretation makes the diagnosis of OMI 50 minutes sooner than STEMI criteria in this case. In our recent external validation, the average time savings compared to STEMI criteria was about 3 hours. Read for yourself here.
2. Hyperacute T waves are now endorsed by the ACC as a full STEMI equivalent finding (though they do not give any objective criteria for how to diagnose them).
3. STEMI criteria are often met when there is no OMI (false positives). In our prehospital study of 117 cases, STEMI criteria diagnosed only 70% of 48 true OMI and had 15 false positives.
Register for access to Queen of Hearts here
MY Comment, by KEN GRAUER, MD (4/21/2024):
- As per Dr. Meyers' detailed description — today's initial ECG, in association with the history of new CP — is readily diagnostic of acute infero-postero OMI, with need for prompt cath with PCI.
- In a patient with new CP — the ST-T wave within the RED rectangle in lead III immediately "caught" my eye. Given tiny amplitude of the QRS in lead III — there is no way that the hypervoluminous T wave in this lead (that totally dwarfs the QRS) can can possibly be normal.
- Given this certainty that the ST-T wave in lead III is hyperacute — there is no doubt that the ST-T waves within the BLUE rectangles of the other 2 inferior leads ( = leads II and aVF) — are also hyperacute (obviously disproportionate in lead aVF, being slightly taller than the R wave in this lead, as well as "fat"-at-its-peak and wide-at-its-base — with by extension, less marked but still disproportionate ST-T wave appearance in lead II).
- Confirmation that these hyperacute inferior lead ST-T wave changes are truly hyperacute — is forthcoming from the "magic" mirror-image opposite ST-T wave appearance that we so often allude to between lead III and lead aVL — which is so characteristic of acute inferior OMI.
- As we so often emphasize — normally, there is slight ST elevation with gentle upsloping in leads V2,V3. Loss of this normal appearance in a patient with new CP — suggests acute posterior OMI until you prove otherwise.
- PEARL: Given common blood supply in most patients with acute inferior OMI from either RCA or LCx occlusion — I treasure the finding of abnormal ST-T waves in leads V2, V3 and/or V4 in a patient with suspected inferior OMI as strong support of true coronary occlusion.
- There is no way that the shape of the ST-T wave in lead V3 of Figure-1 is "normal" in a patient with new CP (with the RED rectangle in this lead) — because the ST segment in this lead is flat and there is no ST elevation.
- Knowing that the ST-T wave in lead V3 is definitely abnormal in a patient with new CP — I immediately direct my attention to the ST-T waves in neighboring leads V2 and V4 — which show within the BLUE rectangles that there is ST straightening with shallow-but-abnormal T inversion in lead V2 — and, in the context of definitely abnormal lead V3 — abnormal ST segment flattening in lead V4.
- The "trained eye" should within less than 5 seconds know that in a patient with new CP — that the ST-T waves within the RED rectangles in Figure-1 almost certainly indicate acute infero-postero OMI.
- Knowing this — it should take no more than 15 additional seconds for the "trained eye" to take in the ST-T waves within the BLUE rectangles — and know that your ECG diagnosis of acute infero-postero OMI has been confirmed. You have within seconds — confirmed the need for prompt cath with PCI.
.png) |
| Figure-1: I’ve labeled the initial ECG in today's case. |





-USE.png)